
PDF
0,42 MB

Heme Insights 7 de maio de 2026_HORIBA
1. Estudo de caso morfológico em que os achados combinados corroboram o diagnóstico de linfoma de Sézary. 2. Visão geral da deficiência de glicose-6-fosfato desidrogenase (G6PD) e seus testes.

► Estudo de Caso de Morfologia
► Glicose-6-Fosfato Desidrogenase
► Quiz
► PDF
Dados do paciente: Homem, 76 anos, em consulta na unidade de dermatologia.
| WBC 11,1* (10^3/mm3) |
| RBC 4,4 (10^6/mm3) |
| HGB 13,6 (g/dL) |
| HCT 41,0 (%) |
| MCV 94,3 (fL) |
| PLT 322 (10^3/mm3) |
Comentário do esfregaço: Presença de uma população com aspecto linfomatoso, frequentemente apresentando núcleo muito denso (às vezes clivado), alta relação núcleo/citoplasma e pouco citoplasma. Resultados da imunofenotipagem de linfócitos: População patológica identificada de linfócitos T: CD3 (positivo), CD4 (positivo), CD7 (negativo), CD2 (negativo), CD158k (positivo).
Os resultados combinados corroboram o diagnóstico de linfoma de Sézary.
Exame de Esfregaço Sanguíneo: O esfregaço sanguíneo mostra vários linfócitos grandes com núcleo cerebriforme ou convoluto e citoplasma basofílico escasso a moderado.
Linfócitos (Normal)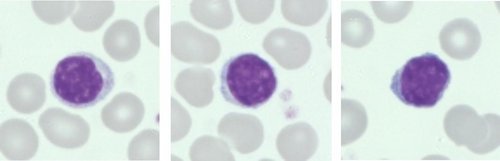
Células de Sézary (Anormais)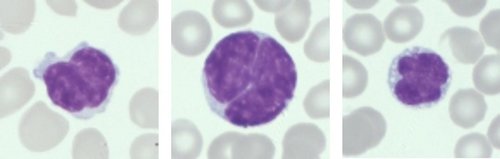
Neutrófilos (Normal)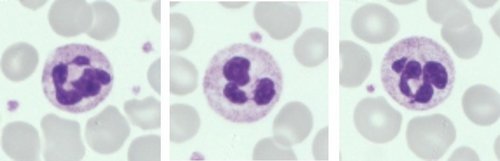
Monócitos (Normais)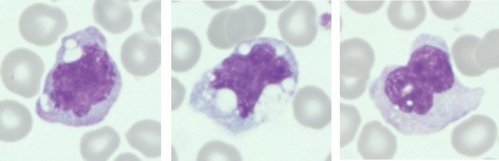
A citometria de fluxo é crucial para um diagnóstico completo e correto.
A síndrome de Sézary é um linfoma cutâneo agressivo de células T que acomete pacientes de meia-idade a idosos e apresenta predominância no sexo masculino. Os pacientes manifestam eritrodermia (vermelhidão afetando mais de 80% da pele), hiperceratose palmar e plantar (espessamento da pele nas solas dos pés), prurido intenso (coceira na pele), alopecia (perda de cabelo), hepatomegalia e linfadenopatia.
A glicose-6-fosfato desidrogenase, ou G6PD, é uma enzima que protege as células contra danos causados pelo estresse oxidativo. A G6PD é a enzima limitante da via das pentoses-fosfato (via das pentoses-fosfato). Nessa via metabólica, a G6PD fornece energia redutora aos glóbulos vermelhos (hemácias) por meio do NADPH e da glutationa (GSSG), protegendo-os do estresse oxidativo que, de outra forma, danificaria a hemoglobina e a membrana eritrocitária. Como as hemácias não possuem mitocôndrias, a única fonte de NADPH é a via das pentoses-fosfato, o que significa que os níveis de G6PD são essenciais para o funcionamento correto dos glóbulos vermelhos. A G6PD é encontrada em todos os organismos, exceto nas arqueas, que são principalmente organismos anaeróbios. A G6PD é expressa em todas as células humanas, indicando seu papel fundamental na proteção celular contra danos (a deleção do gene da G6PD é letal no início da vida embrionária). A atividade da G6PD diminui com o envelhecimento celular, sendo que as células mais maduras apresentam aproximadamente um décimo da atividade original. Os reticulócitos possuem um alto nível de atividade da G6PD (um ponto importante a ser observado na investigação de pacientes com níveis elevados de reticulócitos).
O gene G6PD está localizado no cromossomo X; portanto, os homens possuem apenas uma cópia do gene, enquanto as mulheres possuem duas cópias (ou seja, é ligado ao sexo).
Os homens podem ser hemizigotos para G6PD normal ou hemizigotos para deficiência de G6PD. As mulheres, por outro lado, podem ser homozigotas para G6PD normal, homozigotas para deficiência de G6PD (uma ocorrência rara) ou heterozigotas para deficiência de G6PD. Devido ao processo de inativação ou lionização ligada ao cromossomo X, no qual um dos cromossomos X é silenciado em cada célula, as mulheres heterozigotas apresentam níveis variáveis de atividade da G6PD, desde níveis normais até os esperados em homens hemizigotos.
Existem aproximadamente 230 variantes do gene G6PD com mutações conhecidas, a maioria das quais resulta em diminuição da atividade da G6PD (deficiência de G6PD). O grau de atividade da G6PD varia entre as diferentes mutações. A diminuição da atividade da G6PD provavelmente se deve a um aumento na instabilidade molecular ou a uma diminuição na atividade catalítica, e não a uma diminuição na produção. Devido à natureza da hereditariedade, os homens podem ser portadores tanto de um gene G6PD afetado quanto de um gene G6PD normal. A maior frequência de deficiência de G6PD é encontrada na África Subsaariana, no Oriente Médio e em partes da Ásia.
Deficiência de G6PD
A deficiência de G6PD é a deficiência enzimática mais comum no mundo, afetando aproximadamente 400 milhões de pessoas. A maioria das pessoas com deficiência de G6PD permanece assintomática ao longo da vida; no entanto, anemia hemolítica aguda grave pode se manifestar após a ingestão de feijão, pelo uso de diversos medicamentos comumente prescritos (como primaquina, rasburicase e dapsona) ou mesmo por infecções.
Diferentes variantes de mutação predominam por região: a variante G6PD A− é mais comum na África e nas Américas, enquanto a variante G6PD Mediterrânea é prevalente no Oriente Médio e na bacia do Mediterrâneo. A prevalência da deficiência de G6PD coincide de perto com as regiões onde a malária é (ou era) endêmica, indicando que a deficiência de G6PD oferece uma vantagem seletiva ao proteger contra a malária grave. Células deficientes em G6PD apresentam maior estresse oxidativo, o que dificulta a sobrevivência e a replicação dos parasitas da malária; células deficientes infectadas também são danificadas prematuramente e eliminadas rapidamente durante a passagem pelo baço. A desvantagem disso é que medicamentos antimaláricos comuns, como a primaquina e a tafenoquina, desencadeiam estresse oxidativo, o que pode causar anemia hemolítica com risco de vida se administrados a pacientes com deficiência de G6PD.
Na deficiência de G6PD, a anemia hemolítica é causada quando um agente nocivo (primaquina ou derivados de feijão) gera radicais livres de oxigênio. As hemácias deficientes em G6PD não conseguem detoxificar essas substâncias devido à redução de NADPH e glutationa. Para uma lista dos agentes causadores, consulte a Tabela 1. O dano oxidativo subsequente causa a desnaturação da hemoglobina, formando corpúsculos de Heinz e, consequentemente, células mordidas (hemácias que parecem ter sofrido uma "mordida"; também chamadas de células "mordidas" ou degmócitos), juntamente com danos e rigidez da membrana, remoção esplênica de hemácias danificadas e hemólise intravascular e extravascular. Os pacientes apresentam anemia clínica, icterícia, dor abdominal e, às vezes, febre. O baço pode estar aumentado e a urina geralmente é escura (hemoglobinúria). Um hemograma completo mostrará diminuição na contagem de hemácias e na hemoglobina; LDH e bilirrubina não conjugada estarão elevadas; e a haptoglobina (se dosada) estará diminuída. Deve-se realizar uma contagem de reticulócitos. A preparação de corpúsculos de Heinz pode ser feita corando o sangue com EDTA utilizando um corante supravital, como violeta cristal ou azul cresil brilhante: incubar por 10 a 20 minutos, fazer um esfregaço fino e observar ao microscópio. Os corpúsculos de Heinz se apresentarão como inclusões coradas de roxo próximas à membrana dos eritrócitos. Um teste de Coombs direto deve ser realizado para excluir possível anemia hemolítica autoimune. A avaliação da atividade da G6PD durante o episódio agudo deve ser feita com cautela, pois as células mais deficientes estão sendo substituídas por reticulócitos com maior atividade de G6PD, o que pode gerar resultados falsos "normais". A avaliação da atividade da G6PD deve ser realizada após a resolução da hemólise, preferencialmente de 6 a 8 semanas após o episódio.
Se a anemia for grave, uma transfusão pode ser necessária com urgência; caso contrário, apenas hidratação e analgésicos podem ser suficientes. Se a anemia for relacionada a medicamentos, a suspensão do fármaco é necessária. Observe que alguns medicamentos têm uma meia-vida mais longa do que outros (por exemplo, a tafenoquina tem uma meia-vida muito longa, de 15 a 17 dias, em comparação com 6 horas para a primaquina).
Em casos raros, os pacientes apresentam anemia hemolítica crônica não esferocítica. O quadro clínico é semelhante ao da esferocitose hereditária (por exemplo, icterícia e cálculos biliares), com ampla variação de gravidade, desde leve até grave o suficiente para exigir transfusão. Ao contrário da esferocitose hereditária, o histórico de icterícia neonatal (às vezes grave) é comum.
A Organização Mundial da Saúde (OMS) publicou uma classificação da G6PD relacionada à sua atividade e ao risco de hemólise.
| Classe OMS revisada | Atividade enzimática (% do normal) | Risco basal de hemólise |
| Classe A | < 20% a | Hemólise crônica |
| Classe B | < 45% | Hemólise aguda induzida por gatilho |
| Classe C | > 60% | Sem hemólise |
| U b | Qualquer | Significado clínico incerto |
Uma variante A com atividade <20% será classificada na Classe A somente se estiver associada à anemia hemolítica crônica. Se uma variante com atividade <20% estiver associada à anemia hemolítica aguda induzida por feijão, medicamentos ou infecção, será classificada na Classe B. Se as manifestações clínicas forem desconhecidas, será classificada na Classe U.
b. Atribuição temporária para variantes para as quais atualmente não existem informações suficientes sobre manifestações clínicas.
Essa classificação é usada para esclarecer o regime medicamentoso para pacientes com malária (por exemplo, como mostrado abaixo).
| Recurso | Classe A | Classe B | Classe C |
| Atividade da G6PD | < 20% | 20–60% | > 60% |
| Estágio sanguíneo Tx | ✅ ACT /CQ | ✅ ACT /CQ | ✅ ACT /CQ |
| Primaquina (diariamente) | ❌ | ❌ | ✅ |
| Primaquina (semanal) | ❌ | ✅ (8 semanas) | ❌ |
| Tafenoquina | ❌ | ❌ | ✅ (≥70%) |
| cura radical | ❌ Não é possível | ⚠️ Quantidade limitada | ✅ Padrão |
| Risco de recaída | Alto | Reduzido | Mínimo |
Nota: A cloroquina ou a ACT (por exemplo, artemeter-lumefantrina) podem ser usadas para a Classe A da OMS.
Tabela 1. Medicamentos que podem desencadear anemia hemolítica aguda (AHA) em pacientes com deficiência de G6PD
| Risco definido de AHA | Risco “possível” de AHA |
| Medicamentos antimaláricos | |
| Combinações contendo dapsona | Cloroquina |
| Pamaquina | Quinidina |
| Primaquina | Quinina |
| Tafenoquina | |
| Outras drogas | |
| Ciprofloxacina | Aspirina† |
| Glibenclamida | fosfato de sódio de menadiol |
| Cloreto de metiltionínio | Sulfadiazina |
| Moxifloxacina | Sulfassalazina |
| Ácido nalidíxico | Sulfonilureias |
| Niridazol | Cloreto de tolônio |
| Nitrofurantoína | |
| Norfloxacina | |
| Ofloxacina | |
| Fenazopiridina | |
| Rasburicase e pegloticase | |
| Sulfametoxazol/cotrimoxazol | |
Medição de G6PD
O intervalo de referência recomendado para a atividade da G6PD (medida a 37 °C) é:
12,1 ± 2,09 U/g Hb
351 ± 60,6 U/ 10¹² hemácias
Existem dois tipos de testes para G6PD: qualitativos e quantitativos. Os testes qualitativos (por exemplo, o teste de fluorescência em placa [FST] ou o teste de fluxo lateral) podem fornecer uma decisão rápida sobre se um paciente tem ou não deficiência de G6PD. São relativamente baratos e podem ser realizados rapidamente. Ambos se baseiam na avaliação da produção de NADPH durante um período de tempo definido. Esses testes são adequados para detectar níveis graves de deficiência de G6PD, mas são menos confiáveis em mulheres e durante uma crise hemolítica. Qualquer paciente com deficiência deve realizar um teste quantitativo de G6PD para confirmar o grau de deficiência.
Testes quantitativos, como o reagente Pointe G6PD fornecido pela HORIBA em suas plataformas de bioquímica, medem as concentrações de G6PD. A G6PD catalisa a reação glicose-6-fosfato + NADP⁺ → 6-fosfogluconato + NADPH. Portanto, a produção de NADPH é proporcional à concentração de G6PD, e a taxa de variação da absorbância a 340 nm está relacionada à concentração de G6PD.
Um paciente apresenta hemoglobina baixa, baixa contagem de eritrócitos, volume corpuscular médio normal, mas com contagem de plaquetas muito baixa. O esfregaço sanguíneo mostra a seguinte anormalidade nos eritrócitos e a presença ocasional de eritrócitos nucleados. Qual condição pode estar causando isso?
a) Trombocitopenia microangiopática em conjunto com anemia hemolítica microangiopática
b) Deficiência de ferro
c) Anemia megaloblástica
As respostas serão reveladas duas semanas após o lançamento — confira sua caixa de entrada, nossas redes sociais (LinkedIn / Facebook / X) ou a próxima edição.

A presença de bastonetes de Auer em células blásticas está mais fortemente associada a qual condição?
a) Leucemia linfocítica crônica
b) Leucemia mieloide aguda
c) Anemia por deficiência de ferro
Resposta:b) Leucemia mieloide aguda
As hastes de Auer são inclusões em forma de agulha observadas em blastos mieloides e são características da leucemia mieloide aguda.

Bibliografia
Deficiência de glicose-6-fosfato desidrogenase, blood® 10 DE SETEMBRO DE 2020 | VOLUME 136, NÚMERO 1, Lucio Luzzatto, Mwashungi Ally e Rosario Notaro https://doi.org/10.1182/blood.2019000944
Deficiência de G6PD e testes. https://www.horiba.com/int/healthcare/products/clinical-chemistry/g6pd-deficiency-and-testing/
Equipe Editorial
Kelly Duffy, Andrew Fisher, HORIBA UK Limited

